June 19, 2024 | Uncategorized | Will
Nitric Oxide-mediated Post Translational Modifications (PTM)
Nitric Oxide — a crucial signaling molecule
Nitric oxide (NO) is a highly reactive and rapidly diffusible gaseous molecule with , during which it rapidly breaks down into other nitrogen species. It exhibits a high affinity towards proteins containing heme groups, leading to its canonical activation of soluble guanylate cyclase, which boosts the production of cyclic guanosine monophosphate (cGMP) from cellular guanosine triphosphate (GTP). cGMP, once produced, initiates a multitude of signaling cascades within cells, playing a crucial role in the regulation of various physiological processes. This molecular mechanism serves as the foundation for NO's role as a versatile messenger molecule, regulating diverse biological functions across multiple physiological systems — in the cardiovascular system, it dilates blood vessels and prevents clotting; in the nervous system, it influences synaptic plasticity and memory; within the immune system, it fights microbes and modulates inflammation; in the respiratory system, it dilates airways and regulates breathing; and in the digestive system, it controls gut movements and gastric acid release.
The enzymatic conversion of L-arginine, catalyzed by the primary enzyme nitric oxide synthase (NOS), with the help of the cofactors oxygen, NADPH, and BH4, , leads to the production of nitric oxide and citrulline.
Under normal cellular conditions, the production of NO is tightly regulated. However, in pathological states such as inflammation, oxidative stress, and ischemia, NOS enzymes can overproduce NO, causing nitrosative stress and tissue damage. The excess nitrosative stress can inhibit cell growth, induce apoptosis, and contribute to degenerative diseases, heart failure, cancer, and other pathologies.
The reactivity of free NO radicals make them prone to oxidation and reduction processes, leading to the formation of reactive nitrogen species (RNS) or converting into more reactive forms like peroxynitrite (ONOO-), a potent oxidizing agent. RNS species include nitrogenous products, such as NO• (nitric oxide radical), nitroxyl (HNO/NO−), nitrosonium cation (NO+), higher oxides of nitrogen, S-nitrosothiols (S-NOs), and dinitrosyl iron complexes.
One of the key mechanisms through which NO mediates diverse cellular pathways is by inducing post-translational modifications (PTMs) in proteins. These modifications alter protein structure and function, influencing cellular signaling cascades and physiological responses.
Nitric oxide-induced post-translational changes
NO-mediated PTMs mainly involve three types — metal nitrosylation, protein S-nitrosylation and nitration (Figure 1).
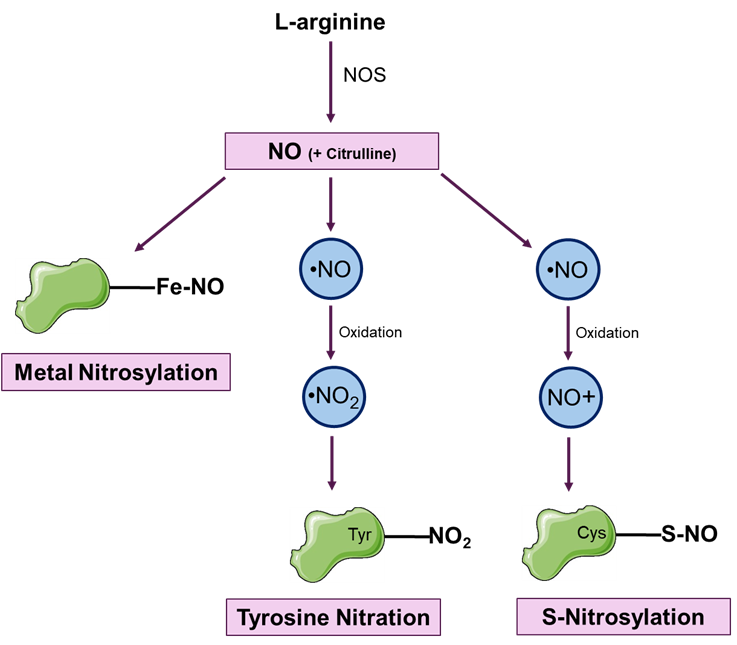
Figure 1: illustrates three types of post-translational modifications (PTMs) mediated by nitric oxide (NO): Metal nitrosylation, where NO binds to transition metals such as the heme-iron (Fe) in proteins; Tyrosine nitration, which involves the addition of a nitro group (-NO2) to tyrosine residues in proteins; and Protein S-nitrosylation, where NO covalently attaches to the thiol groups of cysteine residues in proteins.
Metal nitrosylation
Involves the binding of NO to a transition metal such as the heme-iron (Fe). For example, canonical NO signaling involves the activation of soluble guanylate cyclase by nitrosylation of the iron in its heme moiety, which in turn increases the synthesis of cGMP from cellular GTP. Subsequently, cGMP serves as a crucial second messenger, orchestrating various downstream physiological effects such as smooth muscle relaxation, platelet inhibition, and neurotransmission.
In another example, the interaction of NO with the heme group of cytochrome c oxidase, a crucial component of the electron transport chain, produces the nitrosylated form of the protein. This nitrosylation impairs cytochrome c's ability to accept and donate electrons, leading to a disruption in electron flow within the electron transport chain. Consequently, this disruption affects the overall functioning of the electron transport chain and apoptotic signaling.
Tyrosine nitration
Tyrosine nitration involves the addition of a nitro group (-NO2) to tyrosine residues in proteins, a modification mediated by reactive nitrogen species such as peroxynitrite (ONOO-), nitrogen dioxide (•NO2), and nitronium ion (NO2+). This process occurs under conditions of oxidative and nitrosative stress. While tyrosine is the primary target, other amino acids such as tryptophan, cysteine, and methionine can also undergo nitration due to their susceptibility to RNS. Other molecules susceptible to nitration include lipids, nucleic acids, and small molecules, broadening the impact of nitration in biological systems.
At basal levels, tyrosine nitration serves as a regulatory mechanism in signaling pathways, contributing to cell signaling, apoptosis, and immune responses. However, excessive tyrosine nitration can lead to nitrosooxidative stress, disrupting cellular functions and promoting oxidative damage. This stress arises from the production of reactive intermediates and the inhibition of antioxidant defenses, perpetuating a cycle of oxidative stress and inflammation.
Nitrosooxidative stress induced by tyrosine nitration can modify protein structure and function, altering enzymatic activity, protein-protein interactions, and cellular localization. For instance, nitration of key proteins like cytochrome c, alpha-synuclein, and tyrosine hydroxylase can contribute to neurodegenerative diseases like Parkinson's. Additionally, nitration of tyrosine residues within Aβ peptides can enhance their aggregation and toxicity, contributing to the pathogenesis of Alzheimer's disease. Furthermore, excessive tyrosine nitration has been implicated in cardiovascular diseases, cancer progression, and inflammatory disorders.
Protein S-nitrosylation
Protein S-nitrosylation is perhaps the most prevalent form of NO protein modification. It involves the covalent binding of NO+ to the thiol or sulfhydryl (-SH) group of cysteine residues on proteins. The S—NO bond is formed by NO-derived species, including N2O3, low-molecular-weight nitrosothiols (e.g., S-nitroso-glutathione; GSNO), and S-nitrosylated proteins (transnitrosylation or the transfer of an NO group from one protein to another). Denitrosylase enzymes include the S-nitrosoglutathione reductase system, the thioredoxin system, as well as decomposition mechanisms involving ascorbate and transnitrosylation; together, these systems regulate protein S-NO levels by facilitating the removal of S-nitrosylation from proteins. This reversible PTM is often dependent on the cellular and pathological context of the cell and is a spontaneous reaction mediated by NO. The extent of S-nitrosylation, ranging from mono- to poly-S-nitrosylation, depends on the availability of NO and the biochemical properties of target proteins. Proteomic and immunological techniques have identified over 3000 SNO-modified protein targets, indicative of its widespread regulatory role. However, this catalog may represent only a fraction of the actual targets.
Protein S-nitrosylation holds significant implications across various biological systems, notably in modulating crucial cellular processes and signaling pathways through its influence on protein activity, protein-protein interactions, enzymatic function, and gene expression. The following publications highlight a few specific instances where protein S-nitrosylation plays a pivotal role.
Cardiovascular disease
Sun et al. (2001) examined the impact of S-nitrosylation on the ryanodine receptor (RyR), an essential calcium release channel in cardiac muscle. Their study revealed that NO and similar compounds can S-nitrosylate RyR at Cys-3635, reversing the inhibitory influence of calmodulin on the channel and thereby altering its activity. This modulation of RyR function through S-nitrosylation represents a mechanism through which NO influences calcium signaling in cardiac muscle cells, affecting processes such as excitation-contraction coupling and potentially impacting cardiac function in both healthy and diseased states.
A Closer Look: Cysteine Nitrosylation, RyR2 Channels, and Cognitive Dysfunction
In a recent publication, Dridi et al. (2023) described how cysteine nitrosylation of the ryanodine receptor 2 (RyR2), calcium release channel leads to cognitive dysfunction in heart failure patients. Cognitive dysfunction (CD) is a common complication in heart failure (HF) patients, significantly impacting their quality of life. The underlying mechanisms of CD in HF are complex and multifactorial, involving changes in cerebral blood flow, inflammation, and oxidative stress. One of the key players in this process is the RyR2, a calcium release channel found in the endoplasmic reticulum of neurons. Dysregulation of RyR2 channels has been implicated in various neurological disorders.
Dridi et al. provide significant insights into the role of cysteine nitrosylation, a post-translational modification, in the regulation of RyR2 channels and its implications for CD in HF. The study found that in individuals with HF, hippocampal RyR2 channels exhibited increased cysteine nitrosylation. This modification, along with PKA hyper-phosphorylation and oxidation, leads to the depletion of the stabilizing subunit calstabin2 from RyR2 channels, resulting in their leakiness. This leakiness of RyR2 channels due to cysteine nitrosylation was found to contribute to impaired cognitive function in a mouse model of HF.
Furthermore, the researchers discuss potential therapeutic interventions targeting the stabilization of RyR2 channels. Treatments such as the RyR2 stabilizer beta blocker propranolol, were found to be effective in reducing the leakiness of RyR2 channels caused by cysteine nitrosylation. These findings highlight the critical role of cysteine nitrosylation in the pathophysiology of CD in HF and open new avenues for therapeutic interventions.
Neuronal disease
Cho et al. (2009) studied the effects of S-nitrosylation of dynamin-related protein 1 (Drp1), a crucial player in regulating mitochondrial fission in neurons. They showed that NO, triggered by beta-amyloid protein implicated in Alzheimer's disease, induces mitochondrial fission, synaptic loss, and neuronal damage through S-nitrosylation of Drp1 (forming SNO-Drp1). Mutating Drp1's cysteine to prevent nitrosylation halted these harmful effects. Notably, S-NO-Drp1 levels were elevated in Alzheimer's brains, suggesting its involvement in neurodegeneration's pathogenesis. This elucidates a role for S-nitrosylation role in the neurodegenerative processes of Alzheimer's, linking β-amyloid toxicity, mitochondrial dysfunction, and neuronal injury, with potential therapeutic implications.
Cancer
Marshall & Stamler (2001) highlighted the impact of S-nitrosylation on the NF-κB pathway in cancer cells. They demonstrated that S-nitrosylation inhibits NF-κB activity by modifying a critical thiol in the p50 subunit, preventing its DNA binding, and altering downstream gene expression and cellular responses. This inhibition was reversed by the addition of the denitrosylating agent dithiothreitol, and is modulated by TNFα.The S-nitrosylation-mediated inhibition of NF-κB provides insights into how nitric oxide signaling can influence tumor growth, inflammation, and the immune response within the cancer microenvironment.
Subsequently, a recent review article by Sharma et al. (2021), highlighted the crucial role of dysregulated NO and protein S-nitrosylation in cancer pathogenesis and the tumor microenvironment (TME). Aberrant S-nitrosylation, driven by factors such as altered NOS and denitrosylase expression, oxidative stress, hypoxia, and oncogenic mutations, impacts various cellular processes. A notable example of how oncogenic mutations contribute is the case of p21Ras GTPase. In normal cells, p21Ras, S-nitrosylated at Cys118, facilitates guanine nucleotide exchange (from GDP to GTP) of the catalytic site for enzymatic turnover. However, in cancer cells, p21Ras is often subjected to Gly12Cys and Gly13Cys mutations resulting in S-nitrosylation at additional sites, enhancing its pro-tumor activity. The TME, consisting of diverse cell types and extracellular components, is significantly influenced by S-nitrosylation, which affects immune cells, macrophages, T cells, NK cells, the ECM, and endothelial cells. This modification fosters tumor progression and contributes to therapeutic resistance by enabling cancer cells to reprogram TME components for their advantage.
Diabetes
In a recent publication, Zhou et al. (2023) researchers discovered a significant connection between protein S-nitrosylation and diabetes through the identification of a new enzyme called SCAN (SNO-CoA-assisted nitrosylase). This enzyme utilizes S-nitroso-CoA (SNO-CoA) as a cofactor to S-nitrosylate multiple proteins, including the insulin receptor (INSR) and insulin receptor substrate 1 (IRS1). SCAN's mechanism involves separate domains for SNO-CoA and substrate binding, enabling it to selectively transfer SNO from SNO-CoA to various protein targets. Under normal physiological conditions, SCAN-mediated S-nitrosylation of INSR/IRS1 reduces insulin signaling, serving as a regulatory mechanism. However, in obesity, increased SCAN activity leads to hypernitrosylation of INSR/IRS1, impairing insulin signaling and contributing to insulin resistance, a key feature of type 2 diabetes.
Measuring Nitric Oxide-Induced Protein Modifications
Endogenous levels of S-nitrosylated proteins are typically low, and the S-NO bond is unstable and sensitive to redox changes. Consequently, methods for detecting protein S-nitrosylation can be both limiting and technically challenging. One of the most widely accepted approaches to demonstrate protein S-nitrosylation involves breaking the S—NO bond and detecting the resulting products, particularly free thiols, using techniques such as the biotin switch method.
The biotin switch assay, developed by Jaffrey et al. (2001) is a technique used to detect S-nitrosylated proteins within biological samples. This method involves selectively reducing S-nitrosothiols to expose free thiols, followed by biotinylating the exposed thiols using a thiol-specific biotinylation reagent. This allows the biotinylated proteins to be detected by immunoblot or fluorescent detection techniques.
RayBio® S-Nitrosylation Antibody Arrays
The S-nitrosylation array utilizes a modified biotin switch method to directly visualize S-nitrosylated proteins. With this assay, unmodified free cysteines are initially blocked, and S-nitrosylated cysteines are selectively labeled with biotin-maleimide reagents. The resulting biotinylated thiol groups can be efficiently detected after hybridization with the antibody array.
- Human Protein S-Nitrosylation Array G1 detects S-nitrosylation across 507 proteins in biological fluid samples, while Human Protein S-Nitrosylation Array G2 measures S-nitrosylation across 493 proteins in various biological fluid samples.
- Human Protein S-Nitrosylation Array G8000, our most extensive array, enables measurement across 8000 protein targets, offering unparalleled insight into S-nitrosylation-related protein modifications.
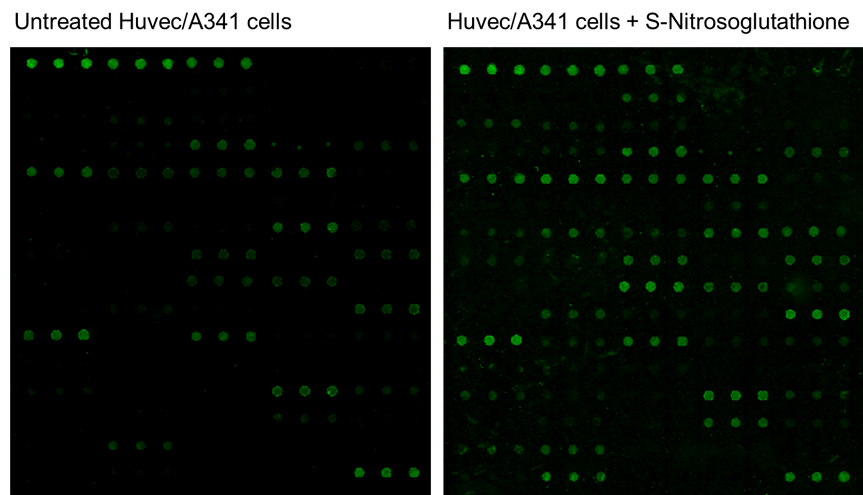
Figure 1: Detection of Protein S-Nitrosylation using the RayBio® Human Receptor Tyrosine Kinase Antibody Array (cat no. AAH-PRTK-G1), which detects 71 human receptor tyrosine kinases. The Biotin-Switch technique was used to covalently attach biotin to previously S-nitrosylated cysteine residues. Left – Untreated Huvec/A431 cell lysates. Right – Huvec/A431 cell lysates treated with 200 µM S-Nitrosoglutathione for 30 minutes at 37°C (to induce Nitrosylation) revealed an increased fluorescent signal intensity of target proteins.
Detecting tyrosine nitration is vital for understanding disease mechanisms. Immunological methods utilizing antibodies against nitrated tyrosine and proteomic-based analyses have been instrumental in detecting and localizing nitrated proteins within cells and tissues.
RayBio® Human Protein Nitration Antibody Arrays
RayBiotech's Human Protein Nitration Antibody Arrays are tailored to detect the relative levels of tyrosine nitration in 507, 493, or 8000 different human proteins in cell lysate. Our 8000-protein array is the most comprehensive offering, providing extensive coverage for in-depth analysis of tyrosine nitration across the human proteome.

Figure 2: Detection of Protein Nitration. Left – Untreated Hela cell lysates were applied to the RayBio® Human Protein Nitration Antibody Array G2 (cat no. AAH-NITRA-G2), which detects tyrosine nitration in 493 human proteins. Right – Hela cell lysates treated with peroxynitrite (to induce nitration) show increased fluorescent signal intensity of target proteins.
Exploring NO signaling and its post-translational modifications is needed to fill critical information gaps in a variety of research and clinical applications. Protein S-nitrosylation and tyrosine nitration, among other modifications, play significant roles in physiological systems and disease pathogenesis. RayBiotech’s range of PTM-profiling tools provides a unique, efficient approach for detecting nitration and nitrosylation, understanding their context, and aiding in the discovery of disease mechanisms and development of potential therapies.
Explore the full range of RayBiotech's comprehensive suite of post-translational modification products. These cutting-edge tools are unique to RayBiotech, and can provide valuable insights into the intricate regulatory mechanisms governing protein function, offering a deeper understanding of cellular processes and disease pathways.
References:
- Cho, D. H., et al. “S-nitrosylation of Drp1 Mediates β-amyloid-related Mitochondrial Fission and Neuronal Injury.” Science, vol. 324, no. 5923, 2009, pp. 102-105.
- Dridi, H., et al. “Heart Failure-Induced Cognitive Dysfunction Is Mediated by Intracellular Ca2+ Leak through Ryanodine Receptor Type 2.” Nature Neuroscience, vol. 26, 2023, pp. 1365-1378.
- Hess, Douglas T., et al. "Protein S-nitrosylation: purview and parameters." Nature Reviews Molecular Cell Biology, vol. 6, no. 2, 2005, pp. 150-166, doi:10.1038/nrm1569.
- Jaffrey, S. R., and S. H. Snyder. "The Biotin Switch Method for the Detection of S-nitrosylated Proteins." Science Signaling, vol. 2001, no. 86, 2001, doi:10.1126/stke.2001.86.pl1.
- Lundberg, J. O., and Weitzberg, E. “Nitric oxide signaling in health and disease.” Cell, vol. 185, no. 16, 2022, pp. 2853-2859.
- Marshall, H. E., and Stamler, J. S. “Inhibition of NF-κB by S-nitrosylation.” Biochemistry, vol. 40, no. 6, 2001, pp. 1688-1693.
- Martínez, M. Carmen, and Ramaroson Andriantsitohaina. "Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease." Antioxidants & Redox Signaling, vol. 11, no. 3, Mar. 2009, pp. 669-702, doi:10.1089/ars.2007.1993.
- Sharma, Vandana, et al. "S-Nitrosylation in Tumor Microenvironment." International Journal of Molecular Sciences, vol. 22, no. 9, 2021, p. 4600, doi:10.3390/ijms220946002.
- Sun, J., et al. “Nitric Oxide, NOC-12, and S-nitrosoglutathione Modulate the Skeletal Muscle Calcium Release Channel/Ryanodine Receptor by Different Mechanisms.” The Journal of Biological Chemistry, vol. 276, no. 26, 2001, pp. 19186-19192.
- Zhang, Yadi, et al. “The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review.” Neurochemical Research, vol. 45, no. 12, 2020, pp. 2815-2827, doi:10.1007/s11064-020-03136-6.
- Zhou, Hua-Lin, et al. "An Enzyme that Selectively S-Nitrosylates Proteins to Regulate Insulin Signaling." Cell, vol. 186, no. 26, 21 Dec. 2023, pp. 5812-5825.e21, doi:10.1016/j.cell.2023.11.009.